
Introduction
Society is increasingly expecting better performance from the world’s healthcare systems, public or private. This translates into a demand for better access to more therapeutically effective care at the lowest price possible, with little tolerance for any negative outcomes, especially by the world’s regulators and politicians.
Pharmacological supply chains within the industry — including those of drug owners as well as developers; contract research, development and manufacturing organizations; equipment suppliers and more — have ambitious agendas to successfully meet and profitably deliver the social demand for more healthcare value per fixed dollar spent through innovation.
Innovation FEATURE PART 1
Automating Drug Discovery’s Next Breakthrough
Contemporary, successful drug innovation springs fundamentally from a well-managed R&D effort, one increasingly reliant on automation, robotics and high-throughput analytical tools to identify the highest-potential targets quickly and efficiently.
Drug innovators’ ability to create novel life-changing pharmaceuticals relies heavily on a diligent, sophisticated and multidisciplinary discovery process. Scientific advancements in the understanding of the human body and diseases, along with continued adoption of breakthrough technologies like high-throughput screens (HTS), have dramatically transformed the landscape of modern drug discovery. Novel science and technologies are constantly reshaping this field with exciting, innovative research ideas and discovery tools.
High-Throughput Screens and Laboratory Automation
Drug discovery starts by selecting a validated biological target, typically a gene or a protein underlying the disease being studied. The groundbreaking work in understanding the pathogenesis of diseases at a molecular level is often accomplished via academic research laboratories and then published to enrich public scientific knowledge.
Once a target is validated, the search begins for a lead compound — either an organic compound or other drug molecule — which can interact with the target and modify its function. Ideally, the lead molecule will alter the disease course without affecting any off-target molecules. This process involves the generation of lead compounds and cycles of lead optimization, pharmacokinetic profiling and toxicity testing.1
High-throughput screens, a critical element of modern drug discovery, are playing a major role in identifying lead compounds. Introduced to the discipline in the mid-1980s, this innovative technology was expanded significantly in the 1990s with an array of technological innovations that allow HTS to screen a large number of compounds (millions) against the drug target in a timely and cost-effective manner.
This array of innovation includes parallel synthesis/combinatorial chemistry (the technique for rapid generation of every possible variant of a parent compound, physically or virtually); automated high-performance liquid chromatography to purify products of combinatorial synthesis; and especially lab automation to improve the efficiency of HTS and streamline the drug-discovery workflow.
With the aid of automated technology and equipment, screening millions of compounds for leads becomes attainable and economical. Lab automation accelerates the speed of performing large-scale sample analysis with a high degree of reproducibility and accuracy, as well as eliminating some of the tedium of manual lab work. A broad range of routine laboratory procedures, such as chromatography, mass spectrometry, and DNA and peptide synthesis, can be conducted by semi- or fully automated instruments.2
In addition, automation is critical in achieving assay miniaturization. This has become an important feature of HTS in response to the increasing number of chemical compounds and molecular targets. Miniaturized assays, such as microarrays, shorten the throughput screen time by using small volumes of samples and reagents, while improving screen efficiency and reducing costs.
This technology requires precise liquid handling within the range of microliters or even nanoliters. Through the facility of automation, such small-quantity liquid measurement and dispensing can be accomplished by advanced liquid-handling instruments using robot arms. Some of these instruments offer real-time dispense verification and independent pipetting control, ensuring precise and accurate liquid dispensing. They can also function as an integral part of a fully automated system.3
An emerging trend in lab automation is to fully automate drug discovery workflows through “robotic researchers” that apply advanced machine learning and artificial intelligence.
All About Eve
An emerging trend in lab automation is to fully automate drug-discovery workflows through “robotic researchers” that apply advanced machine learning and artificial intelligence. One such system is Eve, a collection of computers connected to instrument automation. Eve combines three separate parts of the drug-screening pipeline into a systemic and integrated process, thus streamlining compound screening, hit validation and analysis. Eve’s developer, professor Ross King (University of Manchester, U.K.), believes that the tool will have a lasting influence on the efficacy of drug discovery, given its ability to intelligently respond to a hit with instantaneous analysis rather than after the screening.4
The demand for high testing accuracy and reproducibility is the main driver for a robust drug-discovery laboratory automation system market ($4.1 billion in 2014). As well as large testing volumes and new drug-discovery technology adoption, laboratory automation systems cover almost every aspect of the discovery process, including liquid handling, plate readers, dissolution testing, storage retrieval, laboratory information management systems and robotic systems.5
Computer-Aided Drug Discovery/Design (CADD)
In addition to the traditional experimental approach, computer-aided drug discovery (CADD) forms an important branch of modern drug discovery and is broadly used to facilitate and expedite hit identification, lead selection and optimization. Availability of a variety of databases (i.e., chemogenomics, pharmacogenomics, protein data banks and therapeutics target databases), improved computer processing power and information technology are fundamental to the CADD approach.
CADD can be classified into two broad categories. The first is the structural-based design of new molecules centered on the desired binding to a target. This approach applies the 3D structural information of a target (i.e., a protein) to reverse engineer suitable binding molecules through simulated docking. The other is ligand-based design, which focuses on developing new molecules based on known active or inactive ligands against a target through ligand chemical-similarity modeling.6 A variety of algorithms can be used to facilitate these two approaches.
The most common use of CADD is to perform virtual high-throughput screening over simulated compound libraries by structure-based, ligand-based or combined methods.7 Another important application of CADD is de novo drug design, in which novel compounds are developed from starting molecules with demonstrated activity by adding one functional group at a time or piecing together fragments into novel chemical entities using construction algorithms. In addition, comprehensive algorithms have been used to predict a drug’s ADME (absorption, distribution, metabolism and excretion), related properties and potential toxicity. MetaSite software for example (Molecular Discovery Ltd., Middlesex, U.K.), also offers in silico structure modification to improve the metabolism profile of the lead compounds.7

Precision Research Models for Better Outcome Prediction in Humans
One challenge facing drug discovery is to predict an investigational drug’s pharmacological and toxicological behavior in humans based on the results of in vitro and animal testing. To increase effectiveness, drug innovators must improve their ability to predict failure and reject drug candidates as early as possible.
According to a study led by the Director of Economic Analysis at Tufts Center for the Study of Drug Development, Dr. Joseph A. DiMasi, the success rate of a drug from phase I to market approval is approximately 11.83%.8 Unacceptable toxicity, lack of desired result and disappointing pharmacokinetics (i.e., ADME) are the main reasons for drug failure.9 Just a 10% improvement in predicting failures before clinical trials can translate into savings of up to $100 million on development costs.10
To meet this challenge, precision research animal models are created by inserting human genetic components into an animal model or by engrafting human cells, tissue or tumor cells to the immunodeficient mice to mimic human organ systems or diseases.10 Due to their “humanized” features, these models address the species-difference issue that plagues traditional animal models, improving the reliability in predicting human outcomes with respect to effectiveness and safety. Precision research models are commonly employed to mimic an array of human disorders.10 The hPXR/CAR/CYP3A4/2D6/2C9 mouse is currently the most genetically humanized model available, with 33 human genetic counterparts substituting the mouse’s own genes. This model is used in predicting induction and inhibition of human cytochromes and drug-drug interactions.11
Another exciting advancement in this area is the emergence of 3D bioprinted human tissue models. The leading technology, exVive3DTM, was developed by Organovo for preclinical testing and drug discovery research.12 The company’s first commercial product — exVive3D Human Liver Tissue — is generated by depositing groups of patient-derived living cells in precise layers by a 3D printer. One significant advancement offered by the living 3D liver tissue is a longer functional and stable period compared to standard 2D liver cell cultures.13 Unquestionably, the 3D bioprinted human tissue models are powerful research tools to assist in understanding a particular disease and treatment.
References
- Perrior, Trevor. “Overcoming Bottlenecks in Drug Discovery.” Drug Discovery World (2010). Web.
- Gwynne, Peter, Gary Heebner. “Drug Discovery and Biotechnology Trends: Laboratory Automation: Scientists’ Little Helpers.” Science (2004). Web.
- Caliper Life Sciences. Intelligent Multichannel Pipetting Workstations. Cheshire, U.K. Web.
- “Robotic Researchers: The Next Step in A*utomated Drug Development.” pharmaceutical-technology.com. 26 Apr. 2012. Web.
- “Drug Discovery and Diagnostics Encouraging Lab Automation Market Growth.” Technavio. 7 May 2014. Web.
- Katsila, Theodora, Georgios A. Spyroulias, George P. Patrinos, Minos-Timotheos Matsoukas. “Computational Approaches in Target Identification and Drug Discovery.” Computational and Structural Biotechnology Journal 14 (2016): 177-184. Web.
- Silwoski, Gregory, Sandeepkumar Kothiwale, Jens Meiler, Edward W. Lowe. “Computational Methods in Drug Discovery.” Pharmacological Reviews 66.1 (2014): 334-395. Web.
- DiMasi, Joseph A. Cost of Developing a New Drug. Rep. Tufts Center for the Study of Drug Development. 18 Nov. 2014. Web.
- Petrova, Elina. “Innovation in the Pharmaceutical Industry: The Process of Drug Discovery and Development.” International Series in Quantitative Marketing 19-81, Vol. 20. 26 Oct. 2013. Web.
- Thyagarajan, Amar. “The Increasing Impact of Precision Research Models on Drug Discovery and Development.” Drug Discovery World (2015). Web.
- Scheer, Nico, Yury Kapelyukh, Anja Rode, Stefan Oswald, Diana Busch, et al. “Defining Human Pathways of Drug Metabolism In Vivo through the Development of a Multiple Humanized Mouse Model.” Drug Metabolism and Disposition 43.11 (2015): 1679-90. Web.
- Organovo Announces Commercial Release of the exVive3D™ Human Liver Tissue. Organovo. 18 Nov. 2014. Web.
- McCabe, Caitlin. “Can 3-D Printing of Living Tissue Speed Up Drug Development?” Wall Street Journal (2015). Web.
Innovation FEATURE PART 2
New Forms of Outreach and Improving Clinical Trial Recruitment and Data Integrity
As the cost of drug development continues to mount in unison with the demand for safer, more cost-effective drugs, the pharmaceuticals market is in need of innovation in clinical trial management and design to keep pace with the demand for more effective trial data.
There’s no dispute that total life-cycle costs of developing a drug are high; these costs continue even post-approval, with approximately $312 million in R&D costs required to support drug products after they have been accepted into the market.1 With clinical trials being one of the largest contributors to these costs, improving trial efficiency and the quality of the data is a priority for the industry as outsourcing trials and related services has proven valuable.
According to the 2016 Nice Insight Clinical Research Outsourcing Survey, 56% of respondents reported spending more than $50 million annually on outsourcing, with 18% spending in excess of $100 million.2 Additionally, 76% of respondents reported outsourcing clinical trial services.2 However, as clinical trials continue to demand an increasing level of competence, with sponsors turning to CROs for trial expertise, there is a competitive imperative among potential outsourcing partners to provide new, innovative solutions to clinical trial challenges, which continues to trouble the industry. One of the most significant challenges occurs before these trials even begin, when researchers attempt to recruit participants — a process that can become costly and time-consuming.
In 2013, The Pew Internet Project found that 59% of adults in the U.S. use the internet to search for health related topics.
Supplementing Recruitment with Social Reach
Historically, trial recruitment has been a challenge and, despite technology, remains as such. A recent study published in PLOS ONE found that nearly 60% of researchers surveyed failed to meet recruitment targets — a problem that can impact the statistical significance and overall success of a trial — or required additional time to meet these targets, delaying development and increasing cost.3
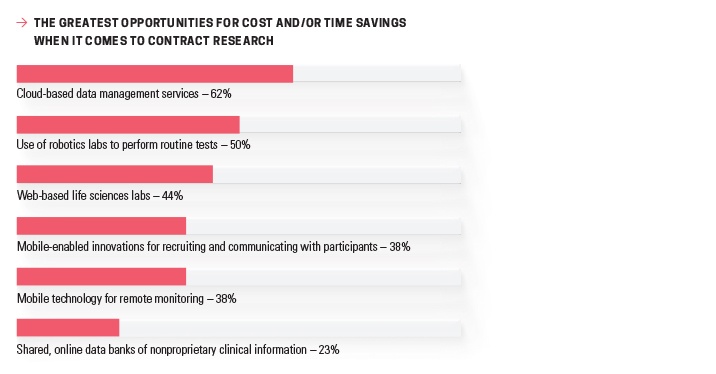
Another review, conducted over eight years, found that “only 31% of multi-center randomized controlled trials achieved their original recruitment targets in a timely manner.”5 Technological advancements can help to improve outreach and engagement in this area, and may help to explain why 38% of respondents felt that mobile-enabled innovations for recruiting and communicating with participants presented a great opportunity for cost and/or time savings in Nice
Insight’s Annual Pharmaceutical and Biotechnology Outsourcing Survey 2015. Just one year later, 48% of respondents reported outsourcing services related to clinical trial recruiting.2,4
The immediacy of communication and the massive reach made possible with social media and digital communication can prove beneficial here, with the PLOS ONE study also finding that supplementing traditional recruitment practices with social media led to a 12-fold increase in recruitment for phase II of the trial in question (as compared to phase I, which only recruited within traditional channels).3 With that, all related social efforts were responsible for approximately 78% of recruitment during the second phase.3
In addition to extensive reach, recruitment via social media is more cost effective in terms of both paid advertising and direct interaction/outreach (i.e., tweeting to followers and/or posting to Facebook/YouTube). When compared to the cost of traditional advertising, which can range from $20 to $500, Facebook ads, for example, typically range from approximately $15 to $20.5 However, if the CRO has an active, respected social media presence, paid advertising may not be necessary; natural engagement is potentially more effective. Despite significant cost and time savings, social forums can also lead to participant issues after a trial is underway.5
Going Social — Open Communications, Open Complaints … and Concerns
Though social media channels may make it easier to recruit and communicate with trial participants, embracing the social landscape can potentially lead to participant issues as well. The growth of the internet as a trusted health information channel has led to the rise of “eParticipants,” or participants that are active on social media during trial participation.6 In 2013, The Pew Internet Project found that 59% of adults in the U.S. use the internet to search for health-related topics. This does not necessarily mean that these interactions are beneficial, however. Social media misuse during clinical trials — including those via social platforms, forums and blogs — can potentially affect the scientific integrity of a study.6 Participants may even disclose sensitive information such as investigational terminology through these interactions, or compare medications with each other.6
The Center for Information and Study on Clinical Research Participation — a nonprofit organization providing clinical trial research information, education and resources to participants and the general public — is attempting to combat these challenges with, in part, a website called “Smart Talking About Clinical Trials.” This initiative aims to educate participants on the risks of openly discussing study specifics by focusing on the influential power of these networks.7 Though organizations such as the Society for Participatory Medicine encourage the involvement and engagement of patients in health decision-making, CROs and sponsors alike must be aware of the risks that this empowerment introduces; additionally, researchers need to be aware of how attempting to monitor such social interactions can jeopardize their own blinding in the trial.6
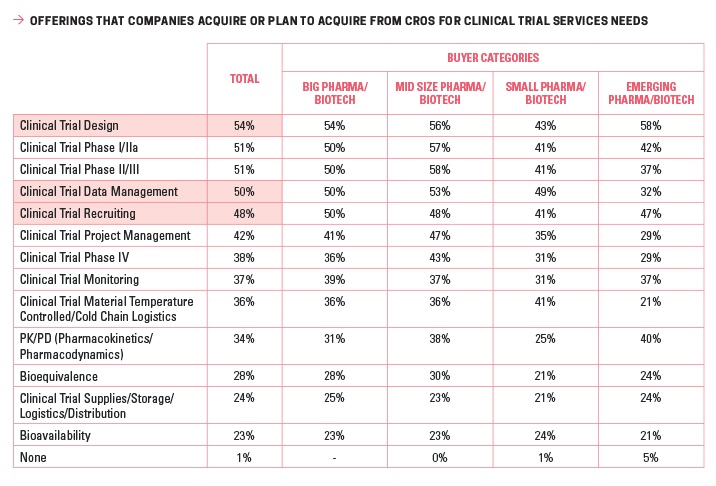
Though research in the area of eParticipants is still relatively limited, and CROs are striving to keep pace with upcoming technology while ensuring that they do not invite unanticipated FDA scrutiny in an area marked by little guidance to date, the industry has much to learn and gain from embracing the social space for recruitment efforts. Engaging an outsourcing partner for everything from clinical trial design to clinical trial data management — outsourced by 54% and 50% of respondents, respectively — may allow sponsors to take advantage of existing expertise that could already include work in the digital space.2
As technology continues to evolve, social platforms grow and system integration becomes deeper across trial sites, sponsors and CROs — an area in which cloud technology will likely play a major role — the digitization of recruitment and other aspects of clinical trials is likely to become a main point of differentiation among CROs looking to innovate in the market. With the term “digital CRO” already appearing and innovation listed as the third-most-important consideration for sponsors considering outsourcing partners, it is likely the move toward social recruitment is already here.2
References
- Tufts CSDD Assessment of Cost to Develop and Win Marketing Approval for a New Drug Now Published. Tufts Center for the Study of Drug Development. 10 Mar. 2016. Web.
- The 2016 Nice Insight Contract Research Organization Outsourcing Survey.
- Shere, Mahvash, Xiu Yan Zhao, Gideon Koren. “The Role of Social Media in Recruiting for Clinical Trials in Pregnancy.” PLoS ONE 9.3 (2014). Web.
- The 2015 Nice Insight Pharmaceutical and Biotechnology Outsourcing Survey.
- Khatri, Chetan, Stephen J. Chapman, James Glasbey, Michael Kelly, Dmitri Nepogodiev, et al. “Social Media and Internet Driven Study Recruitment: Evaluating a New Model for Promoting Collaborator Engagement and Participation.” PLoS ONE 10.3 (2015). Web.
- Lipset, Craig H. “Engage with Research Participants About Social Media.” Nature Medicine 20.3 (2014): 231. Web.
- CISCRP The Centre for Information and Study on Clinical Research Participation. Web.
Innovation FEATURE PART 3
Flexibility in Manufacturing Is Fundamental to Production Success
With record-high drug approval rates, and next-generation therapies that operate via novel mechanisms of action showing great promise in the clinic, the likelihood of innovation seems guaranteed as long as numerous manufacturing challenges are addressed.
Flexibility in all aspects of plant operations, including outsourcing relationships, will be crucial for meeting changing market and regulatory expectations.
Traditional manufacturing strategies won’t allow drug makers to respond quickly enough to increased competition from generics or the entry of new market players, nor reduce development times and costs. With the shift in demand; growth from mature to emerging markets; emerging expectations for local production; a renewed focus on targeted, highly potent therapies; and the ever-greater complexity of new drug substances, fixed, single-product batch manufacturing facilities are no longer relevant for most candidate drugs in the pipeline. Multi-product sites — designed to allow rapid switching between smaller-volume processes for the production of high-quality, high-purity, highly potent, cytotoxic or otherwise challenging and specialized APIs and biologic substances — will be the new norm.

Configurable, Adaptable
State-of-the-art flexible facilities are designed to be configurable and adaptable with more open architecture (facility-wise and equipment-wise to avoid dependence on single suppliers) and extensive automation systems.1 Continuous manufacturing strategies are typically incorporated at some level, and for biopharmaceutical plants, single-use technologies are widely used, although hybrid setups consisting of both disposable and stainless-steel equipment remain common to achieve optimum performance.
For biologics manufacturing, NNE Pharmaplan refers to this approach as the “bio-on-demand” standard.1 Such flexible facilities allow production of different product volumes to meet the needs for both clinical and commercial manufacturing and to rapidly respond to changes in expected market demand (a recent survey of 50 pharmaceutical industry leaders conducted by ORC International, and sponsored by Patheon, revealed that many drug companies over- or underestimate new product demand by up to 25%).2 They also are typically designed to enable rapid switching between different products and product packaging (i.e., vials, cartridges or syringes for parenterals).3
Many Aspects to Flexibility
Multi-product manufacturing and scalability are key features of flexible facilities, as are mobility and replication. Segregation of heating/ventilation/air-
conditioning (HVAC) systems for each production area allows for the manufacture of multiple compounds in a single plant. Continuous manufacturing provides ready scalability from the development lab to the clinic, and on to commercial production. Modular systems provide both mobility and duplicability.
Modular and Flexible
It is important not to confuse modularity with flexibility. Stick-built facilities constructed of modular panels are no more flexible than traditional sites, nor are modular units physically connected in a permanent arrangement that rely on a single HVAC system. Only modular units with individual HVAC systems (autonomous cleanroom POD solutions as referred to by G-CON Manufacturing) that can be readily decontaminated and sanitized for reuse are truly flexible and designed for multi-product processing.4 They are also mobile and can be easily replicated. In addition, because modules are pre-engineered, they can be constructed, installed and commissioned much more rapidly than traditional facilities.
The full benefits of flexible facility designs are only realized with the implementation of appropriately flexible process designs. Modular processing systems and “plug-and-play” equipment provide easy scalability as well as customization for specialized manufacturing and rapid switching of production solutions.5 In bioprocessing, the ability to reconfigure downstream process trains using flexible, portable, disposable units is preferred in multi-product manufacturing sites because often these operations vary significantly for different types of biologics.6

Advances in Automation
When effectively implemented, automation can increase efficiency, productivity and quality while reducing costs. The reason: effective automation requires high-level process understanding in order to identify key areas for both control and optimization.7 Automation is being employed in a variety of applications ranging from batch and recipe management to individual processes to whole-facility automation, including integration of production scheduling and purchasing operations. Connectivity and integration of control systems with different aspects of plant operations allows remote access for monitoring of processes and, when combined with simulation/modeling tools and extensive data collection and analysis, manufacturers are afforded the ability to respond rapidly to process excursions. This also allows for proactive management and exploration of trends for both process optimization and early identification of potential issues.7
Automation is also fundamental to the integration of individual unit operations based on disposable technologies for flexible and continuous manufacturing. In lieu of this need, GE Healthcare’s Life Sciences business and Emerson Process Management announced in late 2015 that Emerson’s DeltaV distributed control system would be incorporated in GE’s FlexFactory integrated biomanufacturing platform based on single-use technologies. Senior Vice President of Industry Solutions for Emerson Process Management, Jerry Brown, expects that “the collaboration will support more predictable processes that eliminate unnecessary work, which translates into a reduced time to market for our customers.”8
Effective automation requires high-level process understanding in order to identify key areas for both control and optimization.
Progress with PAT
Truly flexible manufacturing requires continual access to real-time process data for greater understanding, ongoing optimization and the ability to rapidly respond when upsets or other unexpected events occur. Process analytical technology (PAT) is essential for achieving true consistency, given the variability always present in pharmaceutical raw materials, equipment and processing conditions.9 Advances in portable, nondestructive analytical technologies (e.g., particle imaging, near-infrared, Raman, mass and Fourier transform infrared spectroscopies, focused-beam reflectance) with applications like PAT are making their way into pharmaceutical manufacturing. Effective PAT implementations can result in increased quality, faster product release, reduced cycle times and lower labor and energy costs.10
PAT, combined with automated control platforms, is also key to successfully implementing fully integrated continuous manufacturing operations.9 Companies like Siemens and Rockwell Automation are focused on developing comprehensive solutions that incorporate both. Rockwell, for instance, has been working with G-Con under a grant from the Defense Advanced Research Projects Agency (DARPA) to build a modular facility for flu vaccine manufacturing that can be rapidly installed and up and running in third-world countries.9
Continual Development of Continuous Solutions
Continuous processes provide the scalability needed for flexible manufacturing. The amount of product produced can be increased or decreased simply by running the processes for longer or shorter periods of time. When microreactors are used, numbering up with parallel systems is another solution for increasing production volumes. There are also additional benefits, including a smaller operating footprint, reduced material and resource consumption, reduced quality control needs, more consistent product quality and reduced out-of-spec material.
In fact, in April 2016, the U.S. National Science and Technology Council (NSTC) listed continuous manufacturing of pharmaceuticals and biopharmaceuticals as manufacturing technology areas of emerging priority.11 Several other U.S. government agencies are involved in projects related to continuous pharmaceutical processing.12 In its December 2015 guidance document, Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base, FDA outlined the work its Emerging Technology Team (ETT) is doing with companies to increase the understanding of continuous manufacturing.13
FDA has also recently approved drugs manufactured via continuous processing, including Jannsen’s Prezista, which previously was produced in a batch manner in April 2016.14 The process was developed in collaboration with researchers at Rutgers University, the University of Puerto Rico and the Engineering Research Center for Structured Organic Particulate Systems (C-SOPS), an academic-industry partnership.14 Janssen Supply Chain (JSC), a subsidiary of Johnson & Johnson, is currently investigating applications of other continuous manufacturing techniques for the production of other products that may provide reduced scale-up times and decreased time to market. In addition, Janssen and Johnson & Johnson aim to “manufacture 70% of highest-volume products using continuous manufacturing within eight years, increase yield by reducing waste by 33%, and reduce manufacturing and testing cycle time by 80%.”15
New approaches to continuous process development are also being evaluated. Rather than focus on pharmacokinetics, researchers at C-SOPS look “at material characterization and how minor changes affect manufacturability as part of a system,” according to Associate Director for Industrial Relations and Business Development Doug Hausner.16 Biopharmaceutical manufacturers are also making significant investments in continuous manufacturing technologies and facilities. Eli Lilly, for instance, is investing €35 million to build a continuous API manufacturing facility at its existing manufacturing site in Kinsale, Cork Country, Ireland. The facility will be used for development and commercialization of Lilly’s late-stage pipeline.17
References
- Guldager, Niels, Gert Moelgaard. “Pharmaceutical Production According to the ‘Bio on Demand’ Concept: Flexible Production with Single-use Technologies.” CPP Chemical Plants & Processes. 6 Feb. 2015. Web.
- Flexible Manufacturing Can Help Pharmaceutical Industry Avoid High Costs of Inaccurate Demand Forecasts. Business Wire. 15 Mar. 2016. Web.
- Stauffer, Rebecca. “Pharma Seeks Long Tail Of Flexible Manufacturing.” Bioprocess Online. 18 May 2016. Web.
- Jornitz, Maik W. “The Need For Flexible Facility Designs.” Pharmaceutical Online. Web.
- Markarian, Jennifer. “Modularity Creates Flexible Manufacturing Systems.” Pharmaceutical Technology. 2 Sept. 2014. Web.
- “Flexible Facilities Roundtable.” American Pharmaceutical Review. 31 May 2015. Web.
- Salo, Marko. “Differentiation Through Automation: Driving Growth with Increased Quality and Efficiency - and Reduced Costs.” American Pharmaceutical Review. 30 Nov. 2015. Web.
- GE Healthcare Life Sciences and Emerson Team Up to Drive Biopharma Manufacturing Efficiency. Emerson. 16 Nov. 2015. Web.
- Santos, Sofia T., Francisca F. Gouveia, Jose C. Menezes. “PAT Paves the Way for Continuous Manufacturing.” Pharmaceutical Technology. 1 Sept. 2015. Web.
- Lustri, Jonathan. “Process Analytical Technology-It’s Not Rocket Science, but It Is Science, Math, Control, and IT.” In Tech Magazine. 2015. Web.
- Advanced Manufacturing: A Snapshot of Priority Technology Areas Across the Federal Government. Rep. Subcommittee for Advanced Manufacturing of The National Science and Technology Council. April 2016. Web.
- Markarian, Jennifer. “US National Science and Technology Council Promotes Advanced Manufacturing.” Pharmaceutical Technology. 20 Apr. 2016. Web.
- Advancement of Emerging Technology Applications to Modernize the Pharmaceutical Manufacturing Base. Guidance for Industry. Rep. U.S. Food and Drug Administration. Dec. 2015. Web.
- “FDA Awards Grants to Expand Continuous Manufacturing.” Pharmaceutical Technology. 20 Oct. 2015. Web.
- Advances in Pharmaceutical Supply Chain: Continuous Manufacturing (CM). Apr. 2016. Web.
- Markarian, Jennifer. “Continuous Manufacturing Eases Scale Up for Solid Dosage.” Pharmaceutical Technology. 2 Apr. 2016. Web.
- Lilly Invests $40 Million in Irish Continuous Manufacturing Facility. BioPharmaDive. 7 Apr. 2016. Web.
Innovation FEATURE PART 4
Distribution Channel Security in Ensuring Drug Supply and Safety
Congress passed the Drug Supply Chain Security Act in 2013. Three years into its phased implementation, industry compliance activity continues, as this benchmark ruling comes due in 2017.
Prior to the passing of the Drug Supply Chain Security Act (DSCSA), drug manufacturers were forced to comply with a patchwork quilt of drug “pedigree” laws that varied from state to state. In 2013, the Healthcare Distribution Alliance (HDA) noted that “18 states had adopted final rules regarding distributor licensing and pedigree requirements, three states had enacted legislation but rules were pending, eight states had enacted legislation, one state had proposed pedigree legislation, and 20 states had no legislation or regulations on the topic.”1 Regardless, the industry urged action because nobody wanted to entertain the possibility of having to comply with 50 different state laws.
Seeking relief, industry groups called for unified, federal-level regulation, which became law in 2013. According to FDA, the DSCSA “outlines critical steps to build an electronic, interoperable system to identify and trace certain prescription drugs as they are distributed in the United States.”2 The FDA said the new system “will enable verification of the legitimacy of the drug product identifier down to the package level; enhance detection and notification of illegitimate products in the drug supply chain; and facilitate more efficient recalls of drug products.”
For the most part, the world’s pharmaceutical regulators have also joined the effort and are reconciling their regulations to improve the ability of the global pharmaceutical industry to create a robust, transparent supply chain that ensures the drug supply is reliable, safe and secure from counterfeiters, or any others looking to profit by exploiting gaps.
Initial FDA guidance promised “ten years after enactment, the system will facilitate the exchange of information at the individual package level about where a drug has been in the supply chain.” For drug producers that means by 2023, every vial, bottle, blister pack, combination inhaler and topical tube must be marked and coded accordingly so it can be tracked through every exchange or transaction along the supply chain and its journey to the consumer.
Serialized products allow for a much faster and more effective response to quality/safety excursions and the ability to efficiently remove products from the market well past the point of sale.
Serialization is Coming … Soon!
The FDA timeline shows the next major deadline facing drug manufacturers is the requirement to serialize all products by November 27, 2017. While the feasibility of this is debatable, there are few real technological barriers to implementing an adequate solution, whether locally or system-wide, across dispersed operations and facilties. In a June 2016 Contract Pharma article — “Is the Industry Rising to the Challenge of Serialization?” — Staffan Widengren of CDMO Recipharm noted that global pharmaceutical companies selling products at a high risk of being counterfeited have implemented a traceable, unique ID on each product pack for years.3 “In all countries, the unique information required for a pack should be printed both in human-readable format and in some form of data matrix or barcode,” said Widengren. “However, while most countries require a GS1 standard solution they can differ from country to country, such as is the case with the linear barcode required in China and the 2D matrix required in Turkey.”
Pharmaceutical companies need to be well on their way towards implementing a standard solution that includes serialization features, such as thermal printing, 2D-matrix code verification, human-readable text, brand-neutral tamper-evident labeling and the ability to create a standard file format for reporting and storing serial numbers.
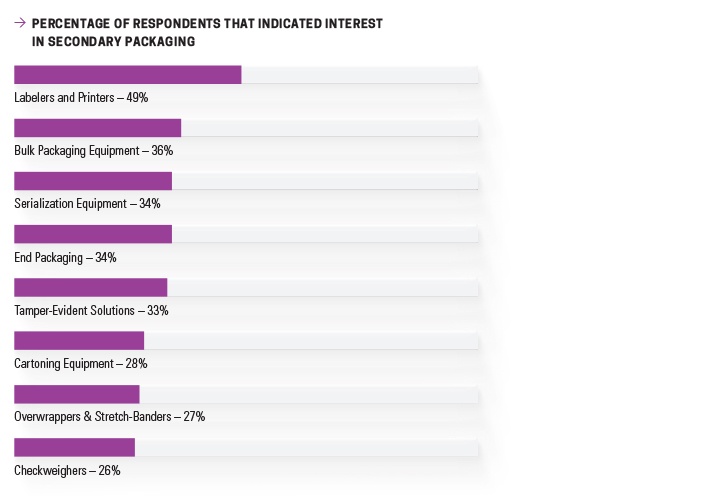
There is evidence this advice is being adopted. Nice Insight’s 2016 Pharmaceutical Equipment Survey found that 49% of those interested in purchasing secondary packaging equipment were seeking to specify labels and printers. New serialization equipment and tamper-evident solutions were among leading technologies being considered for purchase as well, garnering 34% and 33%, respectively.4
According to the Rockwell Automation whitepaper, “Serialization: An Implementation Guide,” a serialization solution should be holistic relative to operations, and enable pharmaceutical manufacturers to comply with current regulations into the future.5 Rockwell’s Global Serialization Lead, Joe Whyte, said the company’s solution is built on industry standards (IEC 61131, ANSI/ISA-88, ANSI/ISA-95) and uses open network and communication protocols, as well as commercial off-the-shelf technologies. An effective solution must provide the required data links and web services to connect serialization data layers to the ERP layer and the supply chain cloud, said Whyte. Rockwell Automation identified five layers based on the enterprise and control system levels of the ISA-95 data model:
Level 0: Printers & vision systems: serialization numbers printed & inspected
Level 1: Unit-level controller & human-machine-interface stations: serialization & aggregation data management per station
Level 2: Line controller: serialization & aggregation data management for the entire packaging line
Level 3: Site server: serialization & aggregation data management for the entire facility
Level 4: Business planning & logistics: serialization interface to enterprise resource planning & manufacturing execution system
Level 5: Supply chain track & trace serialization data event repository
To be effective, a serialization solution requires data input from all layers. Whyte’s paper explained standards such as GS1 for the supply chain and EtherNet/IP for manufacturing, combined with off-the-shelf programmable-logic controllers for data connectivity/data management with packaging machines, printers and marking devices and vision systems, will achieve the integration required to drive better business outcomes.
Providing a high-tech electronic pedigree to all drug products, even on a unit level, can be more than just another expensive regulatory cross to bear. Serialized products allow for a much faster and more effective response to quality/safety excursions and the ability to efficiently remove products from the market well past the point of sale. In fact, industry manufacturing-centric information and control technology suppliers like Rockwell and Emerson maintain that the data generated from tracking and tracing drugs will bring tremendous opportunities for “big” data analysis and allow for much more effective decision-making related to distribution and other metrics that improve strategic performance as well as business outcomes.
Track and trace is poised to completely change the way drugs are manufactured, distributed and sold. These systems will increase transparency and, therefore, accountability of all involved in the supply chain. Track and trace technologies will become an even more critical component as drug developers respond to society’s right to a safe, reliable drug supply.
Innovation Ambition: To Be Continued
To secure an innovative future, drug developers and manufacturers are marshaling their resources and applying technology in ways that might have been unrecognizable 40 years ago. For example, drug discovery pathways are increasingly reliant on automated technology and equipment to screen millions of compounds. Investment here has proven to deliver substantial returns, as lab automation has the ability to accelerate large-scale sample analysis while maintaining the high degree of reproducibility and accuracy demanded by regulators.
Innovation in drug discovery will remain dependent on technological advances like Eve, which combines the drug screening pipeline’s three elements into a systematic, integrated process. Eve and similar technologies will be in high demand to introduce new efficiencies into compound screening, speeding hit validation and enhancing data integrity. Similarly, CADD methods will play an important role in drug innovation supporting structural-based and ligand-based de novo drug design using construction algorithms.
As knowledge of therapeutic value and market potential of compounds or molecules occurs in clinical trials, innovation is paramount in this sector as well. Trial recruitment managers are quickly learning to leverage social media. General access to the internet by consumers allows eParticipants to be active online during trial participation, which allows for a controlled and monitored trial, ultimately producing the data required to prove value.
Future innovations will also be extremely reliant on an elastic supply chain, ready to contract during market adversity or stretch to meet growth in current demand. Here the integration of flexible manufacturing capacity with the latest automation — PAT — and increasingly continuous processing techniques will have an important and lasting impact on the industry’s ability to respond to both consumers’ and regulators’ demands.
Society is demanding so much from the pharmaceutical industry in general, with the pressure to deliver being felt across all sectors of the life sciences and healthcare supply chain.
References
- “Pharmaceutical Traceability.” Healthcare Distribution Alliance. Web.
- “DSCSA Standards for the Interoperable Exchange of Information for Tracing of Certain Human, Finished, Prescription Drugs: How to Exchange Product Tracing Information.” Rpt. in EAS Consulting Group. Silver Spring: U.S. Food and Drug Administration. Web.
- Widengren, Staffan. “Is the Industry Rising to the Challenge of Serialization?” Contract Pharma. 2 June 2016. Web.
- The 2016 Nice Insight Pharmaceutical Equipment Survey.
- Whyte, Joe. “Serialization: An implementation guide.” Rockwell Automation. Nov. 2015. Web.











